Recently, Puhe Biopharma disclosed the development process of its Phase 3 small molecule EGFR inhibitor YK-029A in the European Journal of Medical Chemistry.

Targeting the epidermal growth factor receptor EGFR del19 or L858R Ex21 sensitive driver mutation has achieved great success in the field of NSCLC. Since the first EGFR tyrosine kinase inhibitor (TKI) gefitinib was approved by the FDA in 2003, multiple EGFR inhibitors (first generation: gefitinib and erlotinib, second generation: afatinib and dacomitinib, third generation: osimertinib) have also been approved and have shown good clinical efficacy. Unfortunately, for NSCLC patients with EGFR Ex20ins (EGFREx20ins), the available treatment options are limited, and currently chemotherapy or immunotherapy are mainly used. In addition, patients with sensitive mutations may experience disease recurrence due to acquired T790M resistance when treated with first generation inhibitors, and among patients using first generation inhibitors, T790M resistance accounts for the highest proportion. Given the enormous clinical unmet needs faced by NSCLC patients with EGFR mutations, Puhe Biopharma hopes to find a new molecule to simultaneously address both of these challenges.
The T790M, also known as gatekeeper mutation, causes a decrease in activity towards gefitinib (or erlotinib) due to two reasons: 1) The side chains of methionine residues undergo steric hindrance with inhibitors; 2) The T790M (del19/T790M or L858R/T790M) mutant significantly increases its affinity for ATP. The co mutation of T790M is also the main resistance mechanism of second-generation irreversible TKIs (such as afatinib). Recently, Osimertinib has been found to effectively inhibit tumor growth in patients with T790M co mutation, with a progression free survival (PFS) of 12.3 months. In addition, Almonertinib and Furmonertinib have also been approved in China for the treatment of advanced NSCLC patients with T790M co mutation.
EGFR Ex20 (amino acid insertion positions 762-774) is located at the C-helix of EGFR, and approximately 4-7% of NSCLC patients contain Ex20ins mutations, including dozens of insertion types including but not limited to A763_Y764insFQEA, V769_D770insASV, D770_N771insSVD, D770_N771insNPG and H773_V774ins_NPH, etc. Therefore, the structural analysis of most Ex20ins is missing. In 2013, Yasuda et al. reported the crystal structure of EGFR D770_N771insNPG mutation was observed, and it was found that the insertion of NPG resulted in sustained activation of EGFR, but did not reduce its affinity with ATP. In contrast, FQEA is inserted into A736_Y764, an additional spiral structure was formed, causing adjacent residues to move towards the N-terminus and stabilizing the water transport around L858, thereby keeping the protein in an inactive state. Unfortunately, due to the lack of sufficient safe treatment windows, almost all traditional EGFR inhibitors are ineffective against Ex20ins mutations. Recently, the EGFR-MET dual antibody: amivantamab, has been approved by the FDA for the treatment of second-line EGFREx20ins patients with platinum resistance; Mobocerinib (TAK-788; Figure 1) has also been approved by the FDA for its excellent ORR (43%) in treating second-line EGFREx20ins. However, there is currently no effective treatment for EGFREx20ins in first-line patients. The new generation EGFR inhibitor YK-029A of Puhe Biopharma is currently undergoing a Phase III clinical trial of head-to-head platinum-containing chemotherapy in first-line EGFR Ex20ins patients.
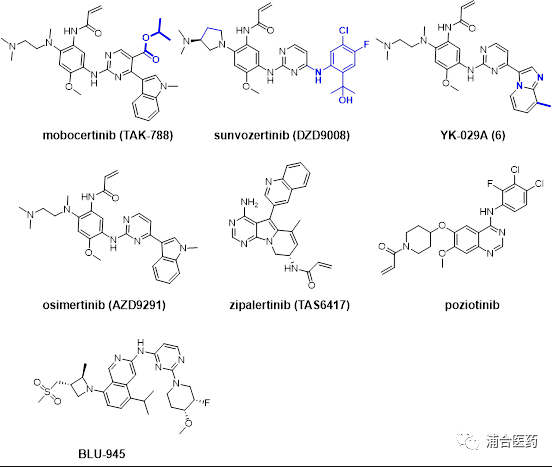
Figure 1. Representative EGFR inhibitor structure
The biggest challenge in designing targeted EGFREx20ins is the selectivity for wild-type EGFR (wt EGFR). Sensitive mutations (del19 or L858R) significantly reduce ATP binding affinity compared with wt-EGFR; The EGFREx20ins mutation increased the affinity with ATP. Therefore, the increase in the activity of Ex20ins mutations usually leads to a decrease in selectivity and loss of the treatment window. The inhibition of off target wild-type EGFR can bring some side effects in clinical practice, such as rash and diarrhea. The most common side effects of using 160 mg mobocerinib include (TRAEs; 30%): diarrhea (90%), rash (45%), and paronychia (34%), with a 17% discontinuation rate.
In clinical practice, it has been found that some EGFREx20ins patients can demonstrate some clinical effects by using higher doses of Osimertinib (160 mg) through off-lable therapy (recommended dose of Osimertinib 80 mg), but it also brings more toxic side effects. Therefore, we realize that increasing its activity and selectivity for Ex20ins mutations may lead to better clinical treatment outcomes. Phe723 is a conserved amino acid in the P-loop, and whether it is T790M, wt (Figure 2), or even the Ex20ins, it is located at the same position. Therefore, we hope to improve the reversible binding affinity for EGFR (mutant or wt) to achieve the final activity enhancement. Since the amino acid sequence of EGFREx20ins in the kinase domain ATP binding region is completely consistent with the wild-type, we can use wild-type EGFR to simulate EGFREx20ins (Figure 2). In addition, due to the strongest binding affinity between wt EGFR and ATP [Ki=5 μM] by increasing the reversible binding affinity with EGFR, the activity of EGFREx20ins will ultimately increase. Due to the different ATP binding affinity, the molecule can increase activity while maintaining selectivity.
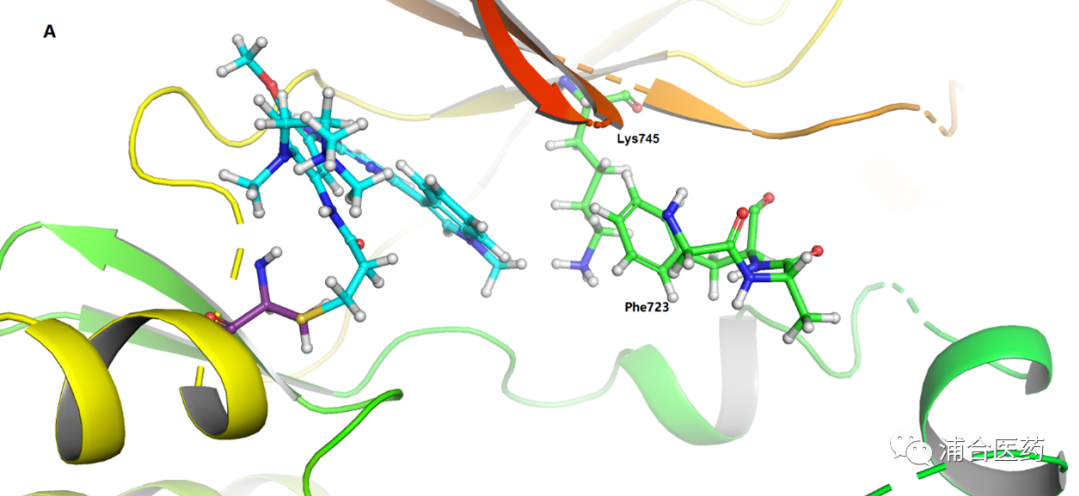

Figure 2A. The X-ray crystal structure of osimertinib bound to the wt-EGFR with 2.31 Å resolution (PDB:6JXT), osimertinib (cyan), Phe723 and Lys745 of EGFR (green); 2B. Surface view (yellow box: hydrophobic pocket near Phe723).
After multiple rounds of optimization and screening, the candidate molecule YK-029A was finally found. The binding mode of T790M is similar to that of EGFREx20ins (Figure S1), and is consistent with the design concept. Through the hydrophobic interaction with Phe723 (Figure 3), the activity is improved and the selectivity is maintained. In addition, there is an unstable (presumably mediated by water molecules) hydrogen bonding with Lys745.
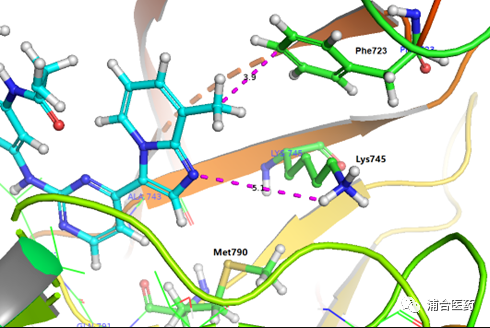
Figure 3. Predicted binding mode of YK-029Ain EGFRLR/TM (PDB:6JX0). Protein: ribbon and green (Phe723, Lys745 and Met790); YK-029A: cyan.
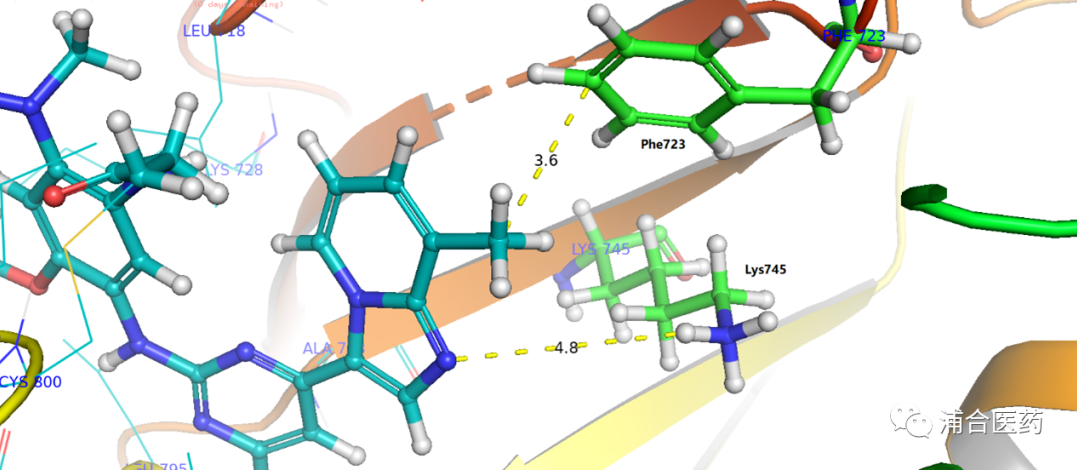
Figure S1. Predicted binding mode of YK-029A in EGFRex20ins (insNPG, PDB:7LGS).
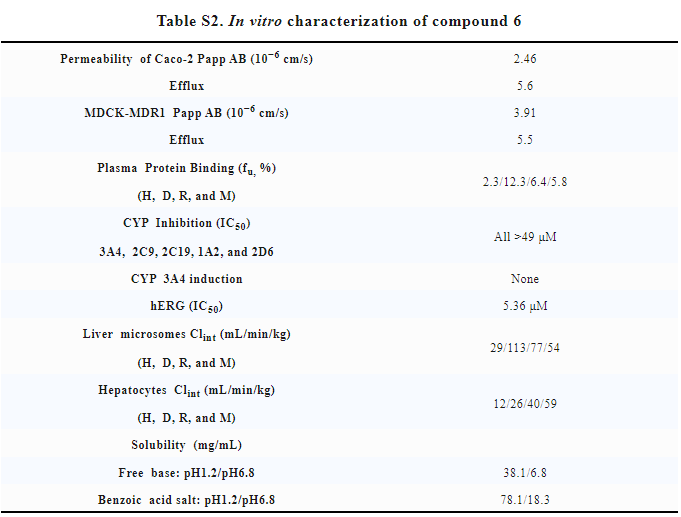
In addition, YK-029A has good in vitro properties (Table S2) without significant inhibitory effect or induction effect on multiple CYP subtypes. Human liver cells have stable metabolism, good solubility and permeability, and good oral bioavailability
In terms of efficacy, YK-029A has good in vivo inhibition effects in both the T790M co mutant CDX model and the Ex20ins PDX model (Figure 5A, 5B) The high-dose group can cause tumor regression without significant toxic side effects, and the weight loss of mice is controlled within 5%. In addition, the targeting effect of YK-029A was once again validated in the PK-PD model (Figures 6A and 6B)
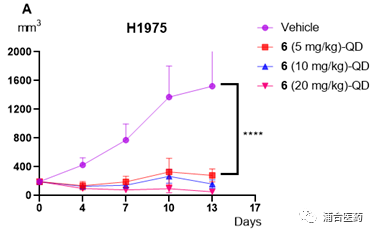
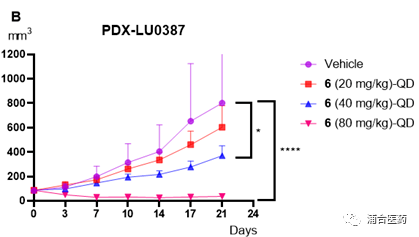
Figure 5A. Antitumor efficacy of compound 6 in the HCC1975 xenograft model of female BALB/c mice (n = 8/group); 5B. Effects of compound 6 in the LU0387 human NSCLC PDX model of BALB/c nude mice (n = 7/group); *p<0.05; **** p<0.0001.
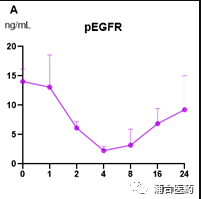
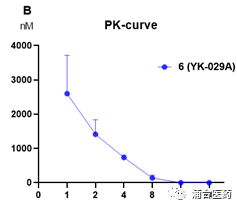
Figure 6A. The pEGFR signal in the LU0387 model after oral administration of compound 6 (80 mg/kg); 6B. The PK curve of compound 6 in the BALB/c nude mice.
In the 28-day GLP toxicology experiment in rats, high-dose YK-029A (80 mg/kg) showed a good safe treatment window, compared to Osimertinib (most rats died at 80 mg/kg). In clinical dose escalation trials, YK-029A at a dose of 200 mg did not show significant serious side effects [low grade diarrhea (≥ Grade 3: 15%) and rash (≥Grade 3: 0%), consistent with the safety observed before clinical use.
Conclusion: Through computer-aided drug design, YK-029A has been identified as a next-generation EGFR inhibitor that can overcome sensitive mutations T790M resistance and EGFR Ex20ins mutations. Due to the lack of precise crystal structure, an unoccupied hydrophobic pocket was discovered through crystal protein structure analysis of Osimertinib and wild-type EGFR. Puhe Biopharma has designed a new generation of irreversible covalent inhibitor YK-029A using this pocket. YK-029A has shown good tumor inhibition effects in EGFRLR/TMCDX and EGFRex20insPDX models in mice. These data support the clinical application of YK-029A, with the aim of benefiting more NSCLC patients. The Phase III clinical trial of YK-029A is currently underway.

Copyright 2025 Suzhou Puhe Biopharma Co., Ltd. 苏 ICP备 2023036064
